|
Wed8 Apr04:25pm(15 mins)
|
Where:
JMS Main Room (438AB)
Session:
Speaker:
|
Trypanosoma brucei, a kinetoplastid parasite, causes severe disease in humans and livestock. Unlike model eukaryotes, its histones are highly divergent and regulate transcription and immune evasion by controlling surface glycoprotein genes. However, how histone sequence variation affects chromatin structure and function has been unclear.
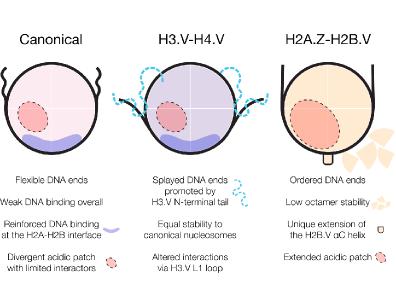
Using cryo-EM, biochemistry, and biophysics, we analysed canonical and variant nucleosomes from T. brucei. We found that canonical nucleosomes are unstable, with weakened DNA binding at entry/exit sites. This instability is compensated by a unique mechanism at the H2A-H2B dimer interface. Additionally, the acidic patch-a major interaction hub in other organisms-is extensively modified in T. brucei, making it resistant to known binders.
Histone variants further reshape nucleosome properties. The repressive variant H3.V, which regulates antigenic variation and transcription termination, causes splaying of nucleosomal DNA ends and lead to more compact chromatin arrays. Activating variants H2A.Z and H2B.V, associated with transcription start sites, substantially remodel the acidic patch, suggesting that interactions with chromatin factors are fine-tuned for transcription control.
By mapping the nucleosome interactome, we further identified variant-specific binding partners that help explain the unusual chromatin biology and virulence of T. brucei.
Our findings highlight how histone divergence underpins unique chromatin architecture in trypanosomes and shed a mechanistic light onto this divergent and clinically relevant pathway to advance neglected tropical disease research.