
|
Poster
40 |
Revisiting quinapyramine: mechanism of uptake, action and resistance |
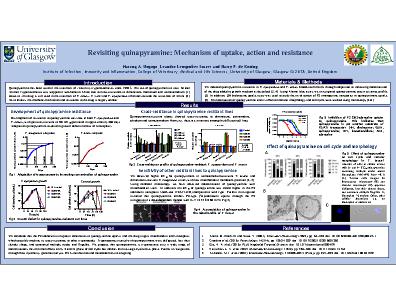
Quinapyramine has been used in the treatment of veterinary trypanosomiases since 1950’s. The use of quinapyramine to treat African Animal Trypanosomiasis was stopped in sub-Saharan Africa due to cross-resistance to diminazene, homidium and isometamidium. However, the drug is still used in the treatment of T. evansi, T. vivax and T. equiperdum infections outside the tsetse belt of Africa. Nevertheless, the mechanism of action and resistance to the drug is largely unclear. We induced quinapyramine resistance in T. equiperdum and T. evansi bloodstream forms through adaptation to increasing concentration of the drug added to growth medium. In vitro development of resistance to quinapyramine was slow in both T. equiperdum and T. evansi, with parasites resistant to 50× EC50 generated in approximately 280 days.Low-level resistant clones, generated after 80 days, did not show cross-resistance to commonly used trypanocides. However, high-level resistant clones, obtained after further adaptation, showed cross resistance to diminazene, pentamidine, ethidium and isometamidium. To investigate the cross-resistance patterns observed, we tested quinapyramine against a panel of drug resistant T. brucei and T. congolense cell lines generated previously. We observed a higher EC50 for quinapyramine in isometamidium-resistant T. brucei and diminazene-resistant T. congolense strains with low mitochondrial membrane potential. Using confocal microscopy, we have observed colocalization of quinapyramine with a mitochondrial stain (Mitotracker). In addition, the EC50 of quinapyramine was 2-fold higher in the P2 adenosine transporter knock-out strain (TbAT1-KO) compared to wild type. Further investigation revealed that quinapyramine inhibits P2-type [3H]-adenosine uptake through the P2 transporter in a dose-dependent manner with Ki = 12. 6 ± 0.7 (n=3). Likewise, an adenosine concentration of 10 µM or higher inhibited the fluorescence-monitored uptake of quinapyramine. The growth of trypanosomes was strongly inhibited in the presence of 5× and 10× EC50 quinapyramine. However, trypanosomal killing was slow, taking 96 – 120 h, compared to diminazene which kills the parasites within 24 – 48 h. In addition, parasite growth recovered when the drug was removed, as exposure for 8 h, 24 h or 48 h did not lead to the death of all the parasites. Cell cycle analysis showed that the majority of cells in cultures treated with 2× and 5× EC50 quinapyramine contained multiple nuclei and/or kinetoplasts from 48 h. Treated cells imaged by fluorescence microscopy and electron microscopy appeared disfigured, lost their slender shape, and contained multiple flagella. Thus, quinapyramine is trypanostatic, has minimal effects on G, S and M phase of cell cycle but inhibits the late-stage cytokinesis phase. We conclude that the P2 adenosine transporter contributes to quinapyramine uptake, and the drug targets mitochondria, which possibly explains its cross-resistance to other trypanocides. Further investigations through genome analysis, RNA-interference and metabolomics are on-going.

